







小林 篤史
1. 動物モデルを利用したヒトプリオン病の病態解析
プリオン病は、すべての健常動物・ヒトに存在する正常型プリオン蛋白が立体構造変化をおこして、感染性をもつ異常型プリオン蛋白(プリオンともよばれます)となり、脳内に蓄積することで引き起こされる神経変性疾患です。ウシの異常型プリオン蛋白はヒトへも感染するため、人獣共通感染症とされています。
ヒトプリオン病の約8割はプリオン蛋白異常化の原因が不明な孤発性クロイツフェルト・ヤコブ病(孤発性CJD)です。孤発性CJDは硬膜移植、下垂体ホルモン投与、脳外科手術器具などを介して伝達されることが分かっており、これらは医原性CJDとよばれています。われわれはこれまでに医原性CJDの動物モデルとして、ヒトプリオン蛋白遺伝子導入マウスに孤発性CJDプリオンを感染させ、その病態解析をおこなってきました。その中で、特定の孤発性CJDプリオン株‐宿主プリオン蛋白遺伝子型の組み合わせは非常に特徴的な病型を示すことを明らかにしました(図1)。参考文献1-3
そして、この特徴的な病型(129M/Mの遺伝子型における中間タイプ異常型PrP蓄積と斑状PrP沈着)が実際にヒトの医原性CJD症例で見られるのか検索したところ、硬膜移植関連CJDの約3分の1はこのような病型を示すことが分かりました(図2)。さらにそれらの症例の実験感染における感染性は孤発性CJDプリオン MV2やVV2と全く同じであることも明らかになりました。これらの結果は、硬膜移植関連CJDの約3分の1が孤発性CJDプリオン MV2やVV2の感染によって引き起こされたことを示唆しています。
(クリックすると拡大します)
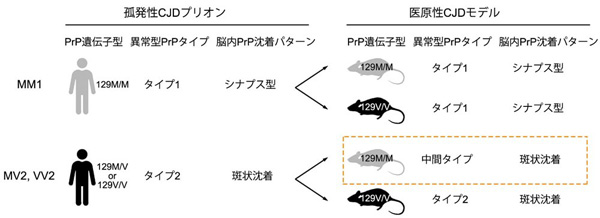
図1.医原性CJDモデルの病型多様性
孤発性CJDプリオンはプリオン蛋白(PrP)遺伝子コドン129の正常多型(メチオニン, Mあるいはバリン, V)と蓄積する異常型PrPのタイプ(1あるいは2)の組み合わせによりいくつかの株に分類される(例: MM1)。それぞれの株は孤発性CJD患者において異なる臨床・病理像をとる。PrP遺伝子コドン129の正常多型は医原性CJDモデルの病型にも影響し、孤発性CJDプリオンMV2やVV2が129M/Mマウスに感染すると異常型PrPのタイプが元のタイプ2から中間タイプに変化する(橙色の枠内)。
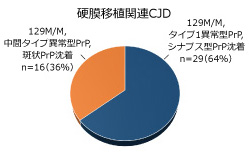
図2.硬膜移植関連CJDの2つのグループ
硬膜移植関連CJDの約3分の1で129M/MのPrP遺伝子型、中間タイプ異常型PrPの蓄積、斑状PrP沈着の組み合わせがみられる。残りの3分の2は129M/Mの遺伝子型で、タイプ1異常型PrPの蓄積とシナプス型沈着を示す。前者は孤発性CJDプリオン MV2やVV2の感染、後者はMM1やMV1の感染で引き起こされたものと考えられる。
その一方、上記の特徴を伴う症例は孤発性CJDにはまったく見つかりませんでした。このことから、上記の特徴的な病型は医原性CJDと孤発性CJDを鑑別する上で重要な手がかりとなると考えられました。そこでこの手がかりをもとに、われわれは過去に孤発性CJDと報告されていた症例を検索し、その中に医原性CJDが含まれていないか検討しました。その結果、2例の医原性CJD(1例は硬膜移植を伴わない脳外科手術歴をもつ症例、もう1例は脳外科医)を同定し、両者において手術器具を介した感染の可能性が強く示唆されました(論文投稿中)。脳外科手術器具を介した感染はこれまで4例しか報告されておらず、1960年代以降は発生していないものと考えられていましたが、われわれの研究成果は脳外科手術器具を介した感染は気づかれていないだけで今でも発生している可能性を示しています。現在、感染性の点でもこれらの医原性CJDを鑑別できるようにするため、簡便な感染性評価法の確立に取り組んでいます。
2. 高齢動物の神経変性疾患の病理学的研究
近年、獣医療・予防医学の発展や飼育管理の向上により、伴侶動物の高齢化が進んでいます。これに伴い、従来はあまり注目されていなかった伴侶動物の認知機能障害・異常行動などが問題になりつつあります。この新たな問題に対し、神経病理学的なアプローチはほとんどされていません。これらの神経変性疾患を正しく診断すること、病理発生メカニズムの解明から治療予防法開発へつなげること、この2点を目指して研究をおこなっていきます。
動物の神経変性疾患は、認知機能の評価が難しく、有用な臨床検査が少ないことから正確な臨床診断が困難です。まず、神経病理学的にこれらの疾患を体系化して分類し、臨床症状・検査所見とのつながりを明らかにすることで、適切な臨床・病理診断基準を確立します。そして、その成果を診療現場に還元し、神経変性疾患の正確な診断に結びつけたいと考えています。
多くの神経変性疾患は、異常な立体構造をとる蛋白質が加齢に伴って蓄積することによって引き起こされている可能性があります。これらの異常蛋白質は自己複製して増殖し、細胞・個体間で伝播しうるという点でプリオンと共通点が多いと考えられます。動物の神経変性疾患を引き起こす異常な蛋白質にはどのようなものが含まれるのか、今後明らかにしていきたいと考えています。
参考文献
| 1. | Kobayashi A, Asano M, Mohri S, Kitamoto T. Cross-sequence transmission of sporadic Creutzfeldt-Jakob disease creates a new prion strain. J Biol Chem. 2007; 282: 30022-30028. |
| 2. | Kobayashi A, Sakuma N, Matsuura Y, Mohri S, Aguzzi A, Kitamoto T. Experimental verification of a traceback phenomenon in prion infection. J Virol. 2010; 84: 3230-3238. |
| 3. | Kobayashi A, Iwasaki S, Otsuka H, Yamada M, Yoshida M, Matsuura Y, Mohri S , Kitamoto T. Deciphering the pathogenesis of sporadic Creutzfeldt-Jakob disease with codon 129 M/V and type 2 abnormal prion protein. Acta Neuropathol Commun. 2013; 1:74. |

